
—— Samarbetsstudie av Zhejiang CDC, Macro & Micro-Test och China CDC Publicerad i Frontiers in Cellular and Infection Microbiology
Studieöversikt
I maj 2026 publicerade Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4.6) en artikel ledd av Zhejiang Provincial Center for Disease Control and Prevention (Zhejiang CDC), med bioinformatikteamet från Beijing Macro & Micro-Test Bio-Tech Co., Ltd. och National Institute for Communicable Disease Control and Prevention (China CDC) som medförfattare. Studien har titeln:
"Identifiering och fylogenetisk analys av sju Brucella abortus-stammar i Zhejiang, Kina."

Denna studie representerar den första systematiska, helgenombaserade fylogenetiska spårbarhetsanalysen av Brucella abortus (B. abortus) i Zhejiang-provinsen, Kina. Teamet analyserade sju isolat som samlats in mellan 2015 och 2025 (fyra stammar av mänskligt ursprung och tre stammar av bovint ursprung från Jinhua, Quzhou och Ningbo). Resultaten ger genomiska bevis för ursprunget och överföringsvägarna för denna "nordliga dominerande art" i en atypisk sydlig epidemisk region i östra Kina.
Bakgrund och betydelse
Brucellos är en zoonotisk sjukdom som orsakas av bakterier av släktet Brucella. Brucella abortus infekterar främst nötkreatur men kan även orsaka sjukdom hos människor. I Kina uppvisar brucellos markant geografisk variation: den högsta incidensen förekommer i norra provinser (t.ex. Inre Mongoliet, Shanxi, Heilongjiang). Däremot har södra provinser, inklusive Zhejiang, historiskt sett dominerats av Brucella melitensis, med mycket få rapporterade fall av B. abortus. Denna regionala skillnad gör genetisk karakterisering och spårning av källan till B. abortus i Zhejiang till en viktig folkhälsoprioritet.
Metoder och viktiga resultat
Forskargruppen antog en mångfacetterad strategi som kombinerar molekylärbiologi och bioinformatik:
1.Patogenidentifiering och grundläggande typning
BCSP-31-gen-PCR och AMOS-PCR bekräftade att alla sju isolaten var B. abortus.
Multilokussekvenstypning (MLST) baserad på nio hushållsgener visade att alla isolat tillhörde sekvenstyp ST2, vilket indikerar hög genetisk homogenitet bland de cirkulerande B. abortus-stammarna i Zhejiang.
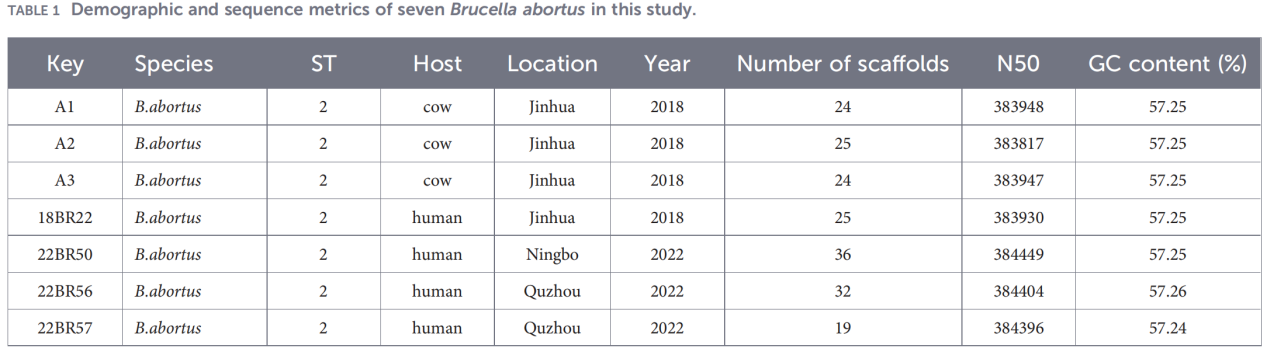
2.Helgenomkarakterisering
Helgenomsekvensering utfördes på Illumina NovaSeq-plattformen. Analys av genomsnittlig nukleotididentitet (ANI) visade att Zhejiang-isolaten hade upp till 99,99 % likhet med referensstammen B. abortus 544.
Pan-genomanalys avslöjade en mycket konserverad population: 3 084 kärngener identifierades, tillsammans med endast 10 skalgener, och inga mjuka kärn- eller molngener detekterades.
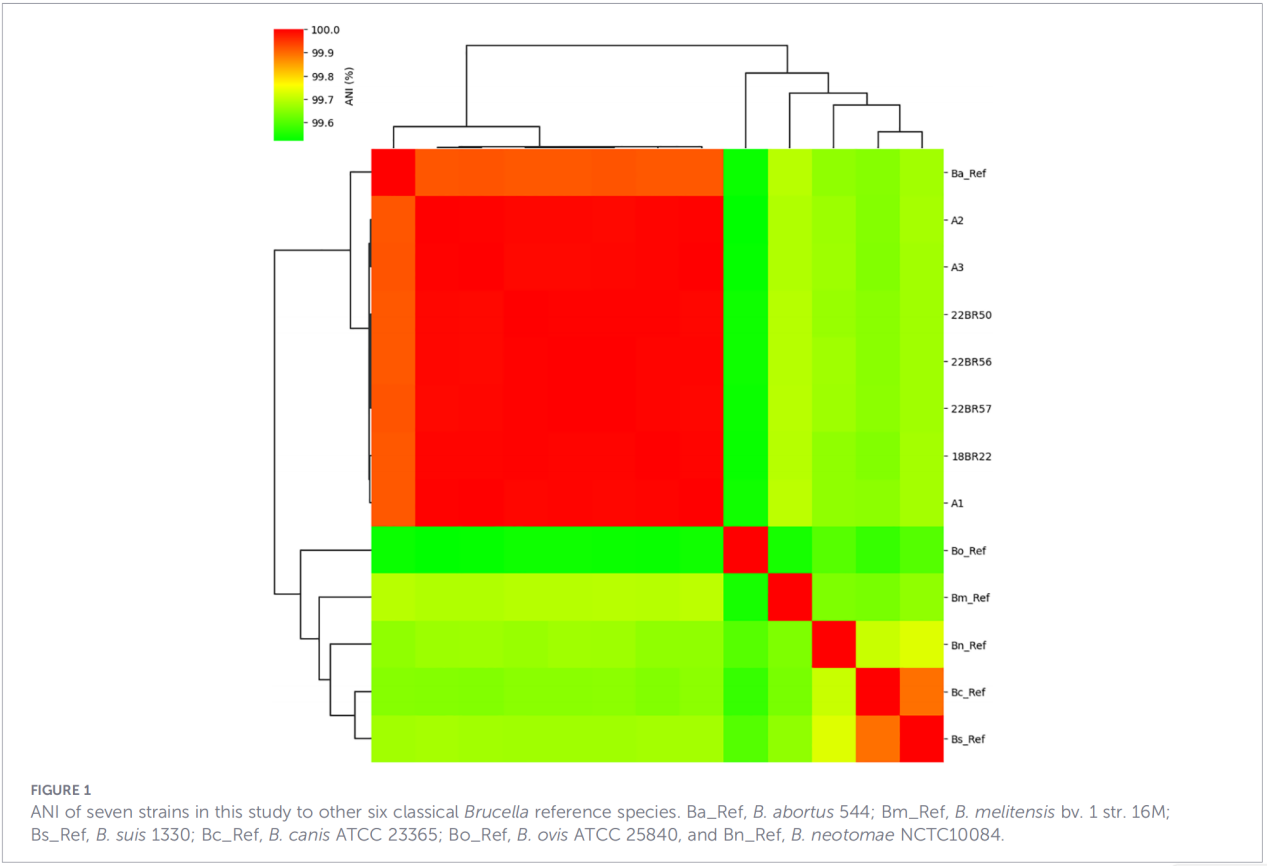
3.Genprofiler för virulens och antimikrobiell resistens
Totalt 68 virulensrelaterade faktorer förutspåddes, vilka täckte klassiska vägar såsom LPS-biosyntes, T4SS-sekretionssystemet och BvrR-BvrS tvåkomponentsregleringssystem. Det är värt att notera att alla isolat saknade adhesin-generna bmaA och btaF. Resistensgenanalys detekterade endast mprF-genen i CARD-databasen, utan några andra resistensdeterminanter identifierades.
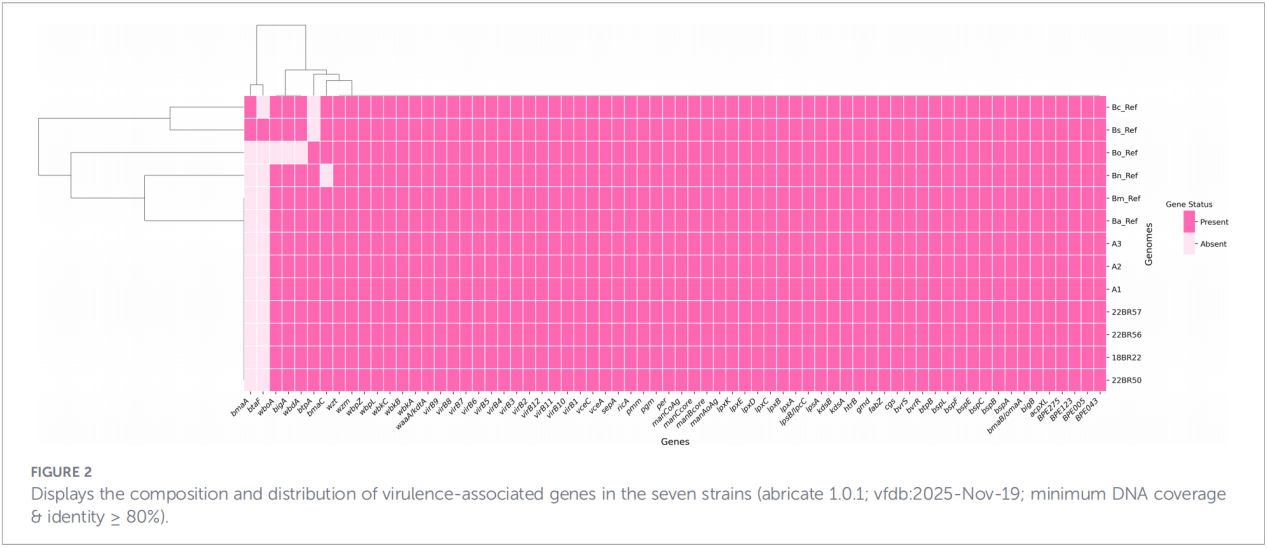 4. Fylogenetisk rekonstruktion och spårning av överföring
4. Fylogenetisk rekonstruktion och spårning av överföring
Analys av kärngenomets enkelnukleotidpolymorfism (cgSNP) placerade Zhejiang-isolaten på en specifik position i det globala fylogenetiska trädet. Resultaten visade att Zhejiang-stammarna bildar en monofyletisk grupp tillsammans med stammar från Ryssland, Mongoliet och flera nordkinesiska provinser (Ningxia, Heilongjiang, Inre Mongoliet, Hebei, Gansu, Peking). Denna grupp delas vidare upp i tre distinkta subklader (klad 1–3), vilket tyder på flera oberoende introduktionshändelser.
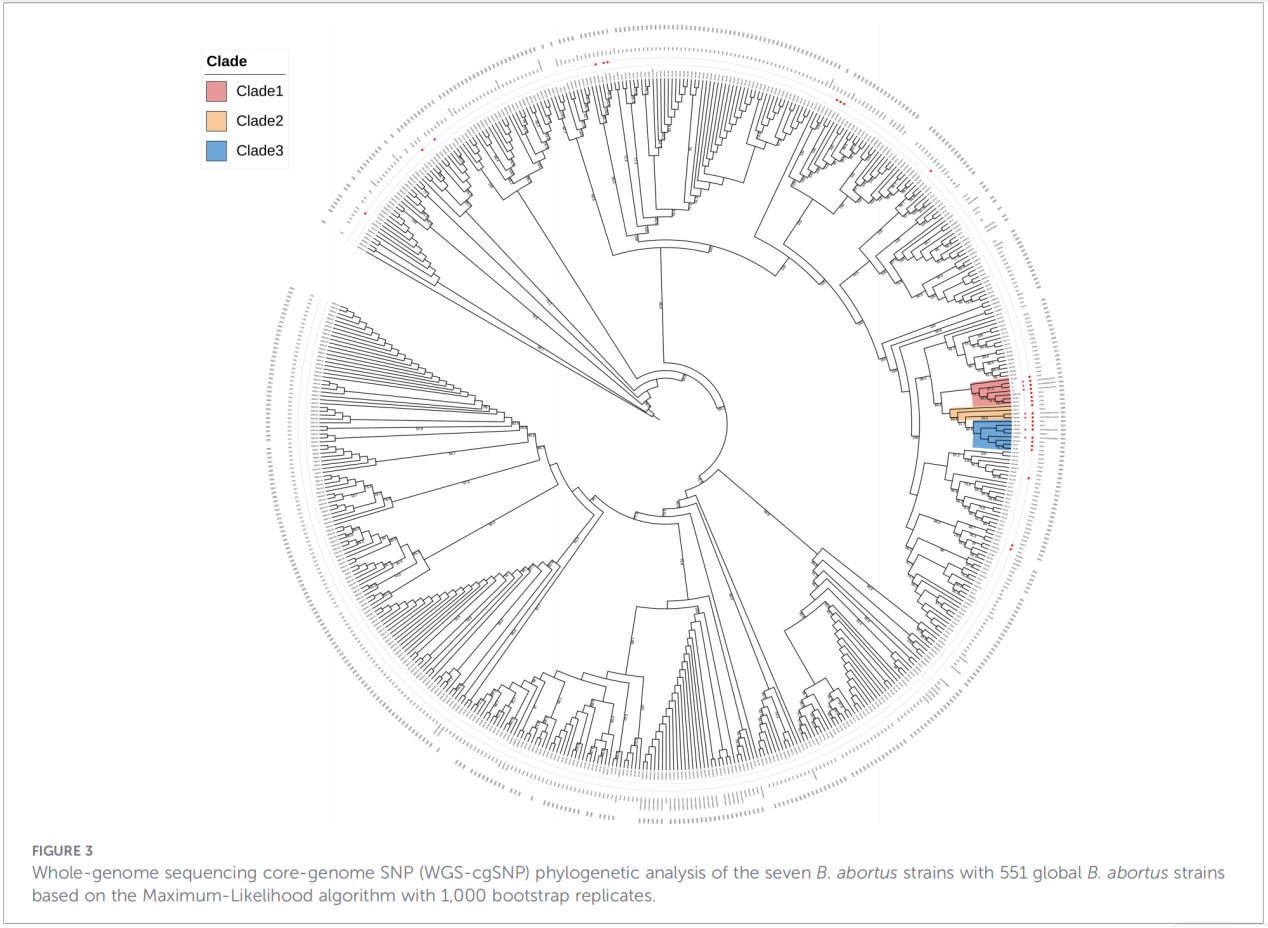
Slutsatser och implikationer
Denna studie tillhandahåller den första högprecisionsgenomiska datamängden för B. abortus i Zhejiang-provinsen och ger flera viktiga slutsatser:
- Clear genetisk bakgrund– B. abortus-stammarna som cirkulerar i Zhejiang tillhör ST2, är genomiskt mycket konserverade och representerar en typisk bovin brucelloshärstamning.
2. Evitätheten av tvärregional överföring– Fylogenetisk analys stöder inte förekomsten av en oberoende endemisk härstamning i Zhejiang. Istället tyder data starkt på att dessa stammar har sitt ursprung i norra Kina och kan dela en gemensam evolutionär bakgrund med stammar från Ryssland och Mongoliet. Förekomsten av tre underklader antyder flera separata introduktionshändelser.
3. Konsekvenser för folkhälsan– Resultaten understryker värdet av genomisk övervakning för brucellos även i traditionellt icke-endemiska regioner som Zhejiang. Även om det nuvarande antalet fall är lågt kan högupplösande verktyg som cgSNP effektivt spåra källan till importerade utbrott och ge vetenskapliga bevis för att avbryta smittspridningskedjor i samband med transport av boskap mellan provinsiella provinser.
Detta arbete fyller inte bara ett forskningsgap i Zhejiang-provinsen utan tillhandahåller också nya baslinjedata för patogenövervakning och riskbedömning av brucellos i Yangtzeflodsdeltatregionen.
Pappersinformation:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Identifiering och fylogenetisk analys av sju Brucella abortus-stammar i Zhejiang, Kina. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Publiceringstid: 10 juni 2026